
Polypeptide Peptides vs Small-Molecule Drugs: What Research on Amlodipine, Prednisone and Metoprolol Reveals About Mechanism Differences
Over 90% of approved drugs on the market today are small molecules — yet peptide-based therapeutics are advancing through clinical pipelines at a faster phase-transition rate than either small molecules or biologics. That contrast raises a precise and important question for researchers: what actually separates these two drug classes at the mechanistic level, and what do familiar drugs like amlodipine, prednisone, and metoprolol teach about those differences?
Understanding polypeptide peptides vs small-molecule drugs is no longer an abstract academic exercise. It shapes how researchers design experiments, select targets, and interpret pharmacological data.
Key Takeaways
- Small molecules like amlodipine, prednisone, and metoprolol are rigid, low-molecular-weight compounds that bind precisely to defined receptor pockets.
- Polypeptide peptides engage broad protein-protein interaction surfaces, functioning more like molecular Velcro than a key-in-lock mechanism.
- Small molecules generally offer oral bioavailability; peptides typically require alternative delivery due to enzymatic degradation.
- Peptides face a conformational entropy cost upon binding that small molecules largely avoid.
- Peptide clinical development is accelerating, with higher phase-1-to-phase-2 success rates than small molecules.
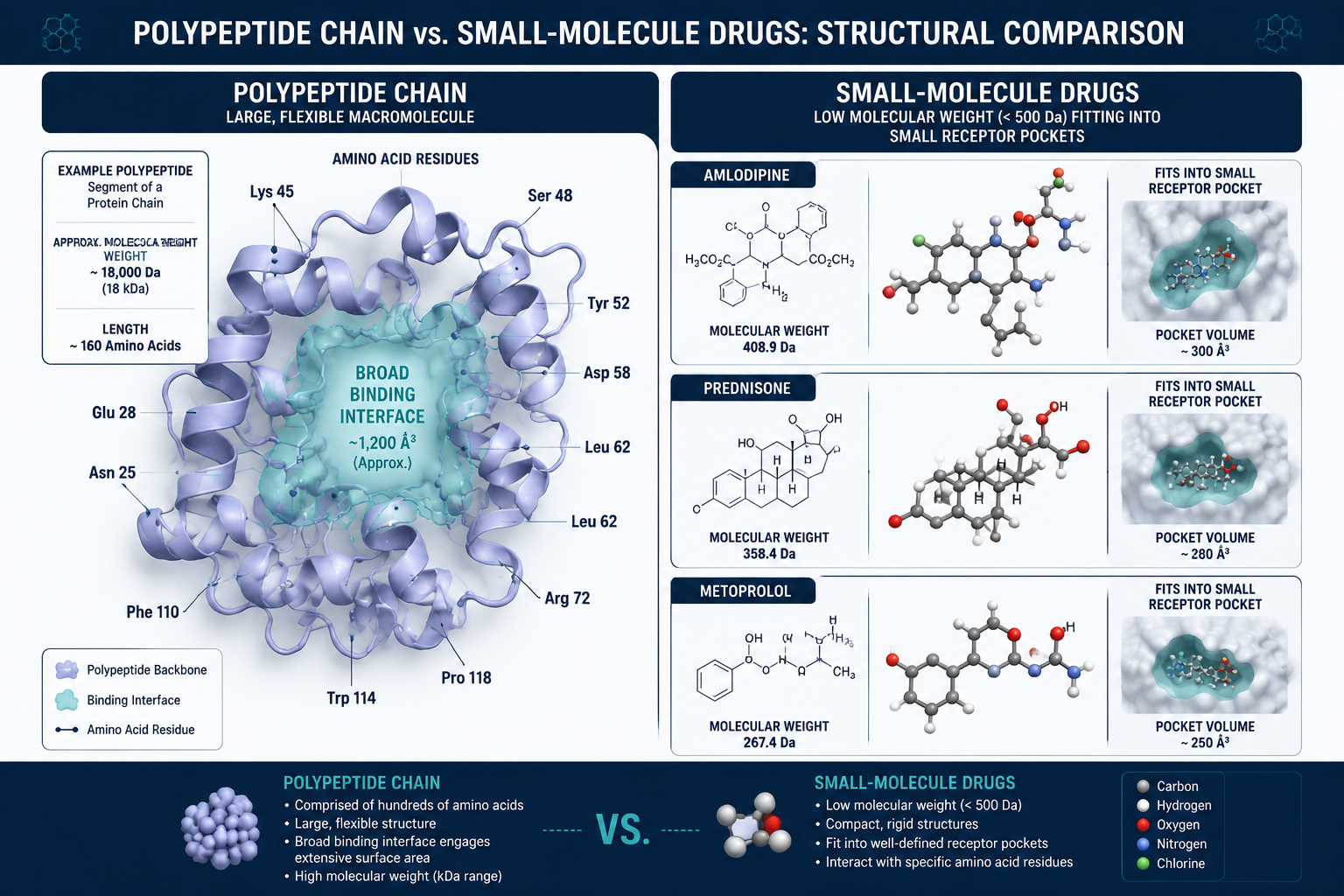
How Small Molecules Work: Lessons From Amlodipine, Prednisone, and Metoprolol
The three drugs most commonly cited in cardiovascular and anti-inflammatory research — amlodipine, prednisone, and metoprolol — are textbook examples of small-molecule pharmacology.
Amlodipine is a calcium channel blocker. It inhibits calcium ion influx into vascular smooth muscle and cardiac cells, producing vasodilation and reduced blood pressure. Its molecular weight sits well under 500 Daltons, and it binds with high precision to a defined pocket on the L-type calcium channel.
Prednisone is a synthetic glucocorticoid. It suppresses inflammation by inhibiting phospholipase A2, cutting off the production of prostaglandins and leukotrienes. Its mechanism depends on entering cells and modulating gene transcription — a task only possible because of its small size and lipophilicity.
Metoprolol selectively blocks beta-1 adrenergic receptors in the heart, reducing heart rate and myocardial contractility. Like the others, it achieves this through enthalpy-driven binding — matching hydrogen bond donors and acceptors within a compact receptor pocket.
"Small molecules derive binding affinity through precise geometric fit — they are rigid keys designed for specific locks."
This precision is their strength. It is also their limitation: small molecules struggle to disrupt large, flat protein-protein interaction (PPI) surfaces where no obvious pocket exists.
Polypeptide Peptides vs Small-Molecule Drugs: Receptor Targeting and Binding Mechanics
Polypeptides — chains of up to 40 amino acids — operate on fundamentally different principles. Rather than fitting into a small binding pocket, they spread across broad molecular interfaces, mimicking the surface of a protein partner. This makes them uniquely suited to disrupting PPIs that small molecules cannot reach.
However, this flexibility carries a cost. Peptides must shed conformational entropy — essentially paying a thermodynamic tax — to adopt the precise active shape required for binding. They exchange that flexibility for enthalpic stabilization upon target engagement. Small molecules, being structurally rigid, largely bypass this penalty.
Research on mitochondria-targeting peptides such as SS-31 (elamipretide) illustrates this well. SS-31 binds cardiolipin on the inner mitochondrial membrane — a large, diffuse lipid surface that no small molecule could engage with equivalent specificity. Explore the SS-31 mitochondrial research themes for a detailed look at this target engagement model.
Similarly, growth hormone secretagogue peptides like those reviewed in tesa peptide benefits research demonstrate how peptides activate receptor cascades through surface-level mimicry rather than pocket occupation.
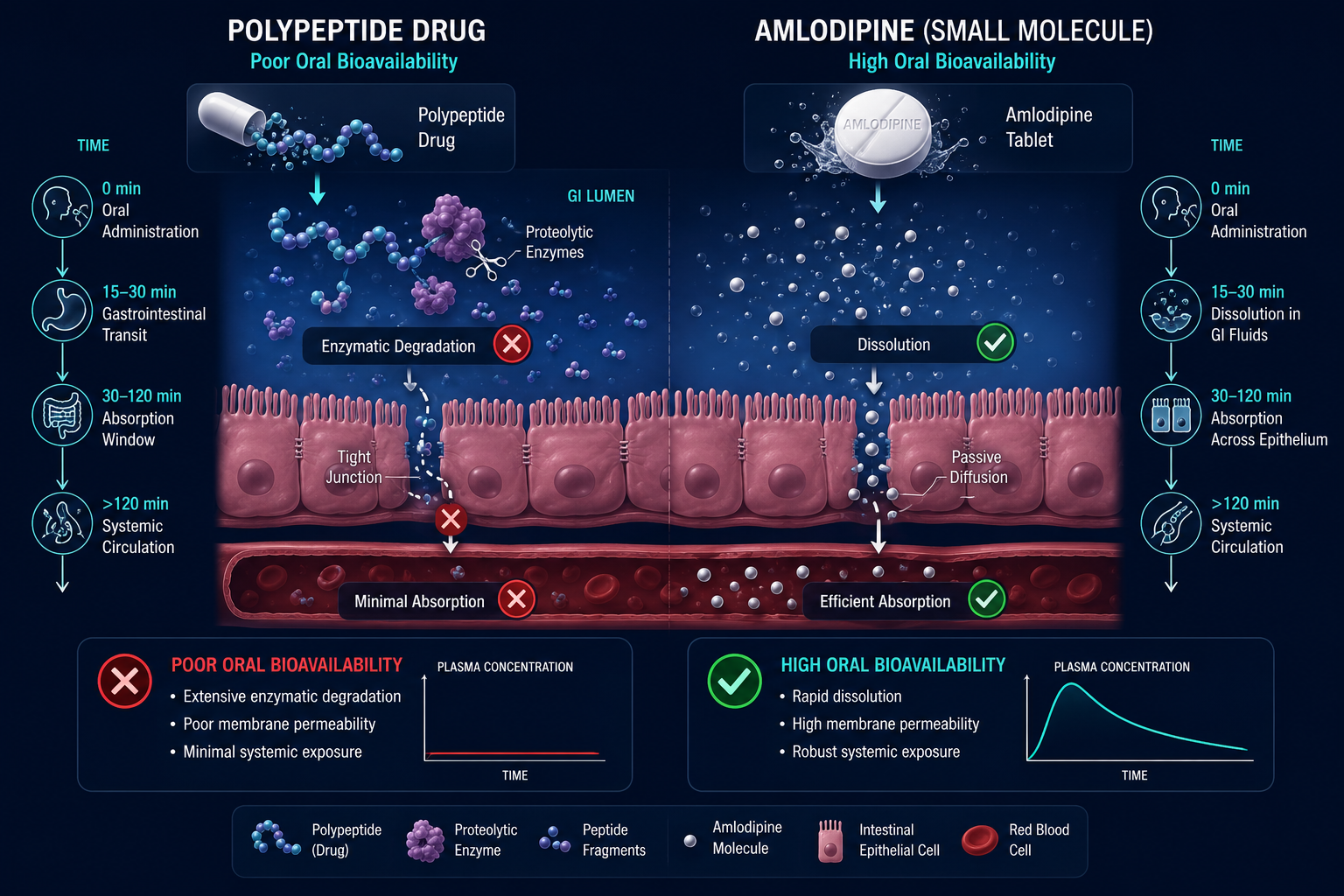
Pharmacokinetics, Half-Life, and Tissue Specificity
This is where the practical gap between drug classes becomes most visible.
| Property | Small Molecules | Polypeptide Peptides |
|---|---|---|
| Oral bioavailability | Generally high | Generally poor |
| Membrane permeability | High (lipophilic) | Low |
| Enzymatic stability | Moderate to high | Susceptible to proteolysis |
| Half-life | Hours to days | Minutes to hours (unmodified) |
| Tissue specificity | Moderate | High (surface-driven) |
Amlodipine, prednisone, and metoprolol are all orally bioavailable precisely because their small size and lipophilicity allow passive diffusion across intestinal membranes. Peptides, by contrast, are broken down by proteases in the gut before reaching systemic circulation, which is why most peptide research protocols involve subcutaneous or intravenous delivery.
Tissue specificity tells a different story. Because peptides engage specific surface architectures, they can be engineered for highly targeted action. Research on MOTS-c metabolic flexibility and GLP-1 incretin research themes demonstrates how peptide ligands can preferentially activate receptors in metabolically relevant tissues with minimal off-target effects.
For researchers exploring peptide half-life optimization, CJC-1295 research themes offer a useful case study in how structural modifications extend plasma stability without sacrificing receptor specificity.
Polypeptide Peptides vs Small-Molecule Drugs: Clinical Trends and Research Implications
The clinical pipeline data reinforces these mechanistic distinctions. Peptides show higher phase-1-to-phase-2 success rates than small molecules, partly because their larger interaction surfaces allow more selective target engagement and a reduced likelihood of off-target toxicity.
Researchers investigating metabolic modulation, tissue repair, or neuroendocrine signaling increasingly look to peptides where small molecules have historically underperformed — particularly at PPI targets. The metabolic modulation research lines overview provides a useful reference for current peptide research directions in this space.
For quality-conscious researchers, ensuring compound integrity is essential. Reviewing quality testing protocols before sourcing any peptide for study is a practical first step.
Conclusion
The comparison of polypeptide peptides vs small-molecule drugs — illustrated through amlodipine, prednisone, and metoprolol — reveals two pharmacological philosophies operating at different scales and surfaces. Small molecules excel at precise, pocket-targeted inhibition with favorable oral pharmacokinetics. Peptides excel at broad surface engagement, PPI disruption, and tissue-selective signaling, at the cost of oral stability.
Actionable next steps for researchers in 2026:
- Map your target: if it presents a defined binding pocket, a small molecule may suffice; if it involves a PPI surface, prioritize peptide candidates.
- Account for delivery route early — peptide studies should plan for non-oral administration from the outset.
- Review half-life data and consider modified analogs for extended in vivo study windows.
- Cross-reference SS-31 dosage and timing research and tesa body composition research themes as model examples of peptide mechanistic study design.
Understanding these distinctions at a mechanistic level is the foundation of rigorous peptide research.